ARGX-113-1801/ADVANCE試験(国際共同第Ⅲ相試験)1-6)
ARGX-113-1801/ADVANCE試験(国際共同第Ⅲ相試験)
「禁忌を含む注意事項等情報」等は電子添文をご参照ください。
試験概要
目的
成人持続性及び慢性ITP患者にウィフガート®を静脈内投与した際の有効性及び安全性を評価する。
デザイン
ランダム化、二重盲検、プラセボ対照、多施設共同(欧州、米国、ロシア、トルコ、ウクライナ、日本)
対象
既存のITP治療で十分な効果が得られなかった又は不耐容であった成人ITP患者131例(日本人8例)
ウィフガート®群:86例(日本人5例)、プラセボ群:45例(日本人3例)
主な選択基準
- ランダム化の3カ月以上前にITPの診断※を受けている。
- スクリーニング時及びベースライン時に測定した血小板数の平均値が30,000/μL未満である。
- 過去にITPに対する治療を1種類以上受けたことがあり、かつ以下のいずれかに該当する。
- 治験実施計画書で規定された併用ITP治療薬を1種類以上、ランダム化の4週間以上前から一定の用量及び投与頻度で受けている。
- 併用ITP治療薬を実施中でない場合、過去にITP治療を2種類以上受けたことがある。
※:ITPの診断は以下により裏付けられた。
(1)米国血液学会(ASH)の診断基準に従った診断が文書で確認され、かつ血小板減少症の他の病因が認められない。
(2)以前にITPに対する治療(TPO-RAを除く)に反応したことがある。
投与方法
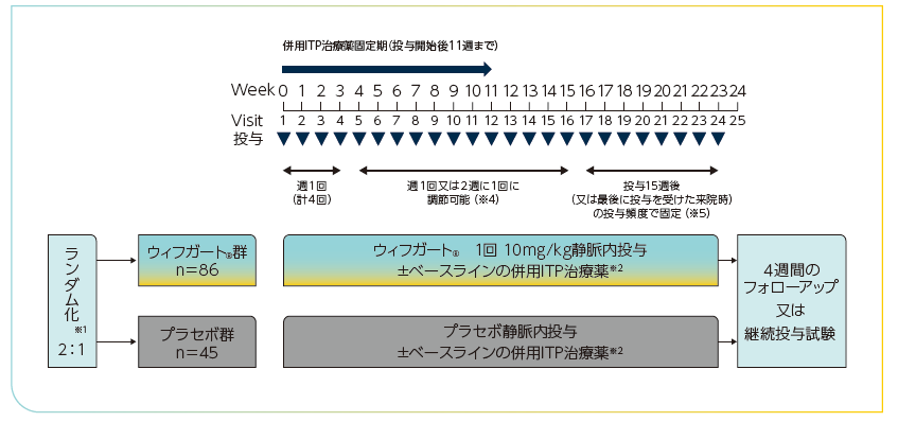
対象患者を2:1の割合でウィフガート®群又はプラセボ群に割り付けた※1。併用ITP治療薬※2及びレスキュー治療※3が許容され、試験中血小板数をモニターした。最初の3週間はウィフガート®10mg/kg又はプラセボを週1回1時間かけて静脈内投与し(計4回)、4~15週は投与頻度を週1回又は2週に1回に調節可能とした※4。16~23週は、投与15週後(又は最後に投与を受けた来院時)の投与頻度で固定した※5。23週の治験薬投与後、1週間の投与終了期間を経て、第Ⅲ相継続投与試験(ARGX-113-1803試験)に移行した。
※1:日本人以外の層別因子:脾臓摘出術の既往あり/なし、ベースラインの併用ITP治療薬あり/なし
※2:併用ITP治療薬(例:経口副腎皮質ステロイド†、経口免疫抑制剤†、ジアフェニルスルホン‡、ダナゾール‡、ホスタマチニブもしくは経口TPO-RA†、又はそれらの組合せ)は、一定の用量及び投与頻度で維持されていることを条件に使用可能とした。なお、ホスタマチニブ及び経口TPO-RAは承認用法・用量に基づく用量調整等が許容された。治験薬投与開始12週後以降、効果不十分(直近4週間の来院での血小板数がいずれも30,000μL未満)な場合は、事前に規定されたITP治療薬の追加又は増量が許容された。
†:ITP治療に対し本邦未承認の薬剤を含む、‡:ITP治療に対し本邦未承認
※3:血小板数が30,000/μL未満で、以下のいずれかに該当する患者に対しては、ベースラインから24週間の治験薬投与期中にレスキュー治療が許可された。
・差し迫った出血リスク又は臨床的に重大な出血又は粘膜出血
・緊急手術の必要性
※4:連続した4回の来院(4回目の来院が判定時の来院)のうち3回で血小板数が100,000/μL以上となり、かつこれら4回の最後の来院時に100,000/μL以上になった場合、又は血小板数が連続した3回の来院で100,000/μL以上になった場合に投与頻度を2週に1回にした。
2週に1回の投与中、連続した2回の来院で血小板数が100,000/μL未満又は1回の来院で血小板数が30,000/μL未満になった場合、又はレスキュー治療を受けた患者は、投与頻度を2週に1回から週1回に増やした。
血小板数が400,000/μLを超えた場合は投与を一時中断し、血小板数が150,000/μL未満に減少したことを確認した上で2週に1回の投与頻度で再開した。
※5:主要評価項目の判定のため、投与頻度の調節は不可とした。
評価項目
【主要評価項目】
持続的血小板数反応※1が認められた慢性ITP患者の割合(検証的な解析項目)
【主要な副次評価項目】 (抜粋)
- 慢性ITP患者における病勢コントロール期間※2
- 全体集団(慢性及び持続性ITP患者)における持続的血小板数反応※1が認められた患者の割合
【その他の副次評価項目】(抜粋)
- 全体集団における各来院時の血小板数のベースラインからの平均変化量
- 全体集団におけるレスキュー治療実施率
【探索的評価項目】
- 全体集団におけるInternational Working Group(IWG)完全奏効※3が認められた患者の割合
- 全体集団におけるIWG奏効※4が認められた患者の割合
- 全体集団におけるIWG初回奏効※5が認められた患者の割合
※1:19~24週の6回の来院のうち4回以上で血小板数50,000/μL以上を達成
※2:規定の24週間の治験薬投与期のうち、血小板数50,000/μL以上となった累積週数
※3:7日間以上間隔を空けた連続する2回以上の来院で、血小板数100,000/μL以上、かつ出血性イベントがない(WHO Grade 0)
※4:7日間以上間隔を空けた連続する2回以上の来院で、血小板数30,000/μL以上、かつ血小板数がベースラインから2倍以上に増加、かつ出血性イベントがない(WHO Grade 0)
※5:4週の時点で、血小板数30,000/μL以上、かつ血小板数がベースラインから2倍以上に増加
解析方法
臨床的有効性の解析は、最大の解析対象集団(FAS:full analysis set)、FAS-慢性ITP、治験実施計画書に適合した解析対象集団(PP:per protocol)及びPP-慢性ITPを対象として実施した。
主要評価項目及び主要な副次評価項目の解析では、全体の第1種過誤確率を制御するため、階層手順にて検定を実施した。各検定の名目上の第1種過誤確率は5%とした。事前に規定した順序で検定を実施し、先に実施したすべての評価項目の解析でP値が0.05未満であった場合にのみ、次の評価項目の検定を実施した。
【主要評価項目】
Cochran-Mantel-Haenszel検定を用いて、ランダム化の層別因子(脾臓摘出術の既往及びベースラインの併用ITP治療薬)並びにベースラインの血小板数カテゴリー(15,000/μL未満 vs 15,000/μL以上)で層別解析を行った。
【副次評価項目】
持続的血小板数反応に関連するすべての副次評価項目は、主要評価項目と同様の解析を実施した。
血小板数のベースラインからの変化量については、mixed models for repeated measures(MMRM)を用いて投与群間の差を解析した。
【部分集団解析】
持続的血小板数反応(19~24週)が認められた患者の割合を、ランダム化の層別因子及び全体集団、ベースラインの血小板数、ITP前治療の数(種類)、診断からの期間、地域、年齢カテゴリー、リツキシマブ前治療及びTPO-RA前治療別に集計した。
患者背景3,5)
| ウィフガート®群 (n=86) | プラセボ群 (n=45) | ||
|---|---|---|---|
| 年齢(歳)、中央値(範囲) | 47.0(19-85) | 55.0(18-82) | |
| 体重、平均値(標準偏差) | 80.2(20.2) | 74.2(17.5) | |
| 性別、例数(%) | 女性 | 47(54.7%) | 24(53.3%) |
| 男性 | 39(45.3%) | 21(46.7%) | |
| 人種、例数(%) | アジア人 | 5(5.8%) | 3(6.7%) |
| 白人 | 80(93.0%) | 41(91.1%) | |
| 報告なし | 0 | 1(2.2%) | |
| その他 | 1(1.2%) | 0 | |
| ベースラインの血小板数/μL、平均値(範囲) | 17,000(0-51,000) | 12,000(2,000-31,000) | |
| 15,000/μL未満、例数(%) | 37(43.0%) | 25(55.6%) | |
| ITPの分類、例数(%) | 持続性ITP(診断から3~12カ月) | 8(9.3%) | 5(11.1%) |
| 慢性ITP(診断から12カ月超) | 78(90.7%) | 40(88.9%) | |
| 最初の診断からの期間(年)、中央値(範囲) | 4.2(0.3-54.1) | 6.1 (0.5-53.4) | |
| ITP前治療薬の数(種類) 中央値(範囲) | 3.0(1-9) | 3.0(1-7) | |
| ITP前治療の数(種類) | 1、例数(%) | 14(16.3%) | 4(8.9%) |
| 2、例数(%) | 13(15.1%) | 12(26.7%) | |
| 3以上、例数(%) | 59(68.6%) | 29(64.4%) | |
| ITP前治療の種類、例数(%) | 副腎皮質ステロイド† | 82(95.3%) | 40(88.9%) |
| 人免疫グロブリン静注又は 抗D人免疫グロブリン静注§ | 42(48.8%) | 29(64.4%) | |
| TPO-RA† | 48(55.8%) | 29(64.4%) | |
| 脾臓摘出術 | 32(37.2%) | 17(37.8%) | |
| リツキシマブ | 31(36.0%) | 14(31.1%) | |
| その他免疫抑制剤† | 21(24.4%) | 18(40.0%) | |
| ホスタマチニブ | 3(3.5%) | 1(2.2%) | |
| ダナゾール‡ | 10(11.6%) | 6(13.3%) | |
| ジアフェニルスルホン‡ | 1(1.2%) | 2(4.4%) | |
| ベースラインの併用ITP治療薬あり、例数(%) | 43(50.0%) | 22(49.8%) | |
| ベースラインの併用ITP治療薬の種類、例数(%) | 副腎皮質ステロイド† | 22(25.6%) | 12(26.7%) |
| 人免疫グロブリン静注又は 抗D人免疫グロブリン静注§ | 2(2.3%) | 1(2.2%) | |
| TPO-RA† | 20(23.3%) | 9(20.0%) | |
| リツキシマブ | 0 | 1(2.2%) | |
| その他免疫抑制剤† | 8(9.3%) | 6(13.3%) | |
| ダナゾール‡ | 2(2.3%) | 1(2.2%) | |
| アスコルビン酸‡ | 0 | 1(2.2%) | |
| 出血性イベント(WHOスケール)Grade 1以上、例数(%) | 42(48.8%) | 29(64.4%) | |
†:ITP治療に対し本邦未承認の薬剤を含む、‡:ITP治療に対し本邦未承認、§:本邦未承認
■治験薬への曝露
ウィフガート®群では86例中15例(17.4%)が投与頻度を週1回から2週に1回に変更しました。投与頻度固定期間(16~23週)に2週に1回の治験薬投与を受けた患者の割合は、ウィフガート®群で67例中10例(14.9%)、プラセボ群で34例中1例(2.9%)でした。
有効性評価
主要評価項目
持続的血小板数反応※1が認められた慢性ITP患者の割合
ウィフガート®群がプラセボ群と比較して統計学的に有意に高く、ウィフガート®群が78例中17例(21.8%)、プラセボ群が40例中2例(5.0%)でした(P=0.0316、Cochran-Mantel-Haenszel検定※2:両側P値)(検証的な解析結果)。
持続的血小板数反応が認められた患者の割合(慢性ITP患者)
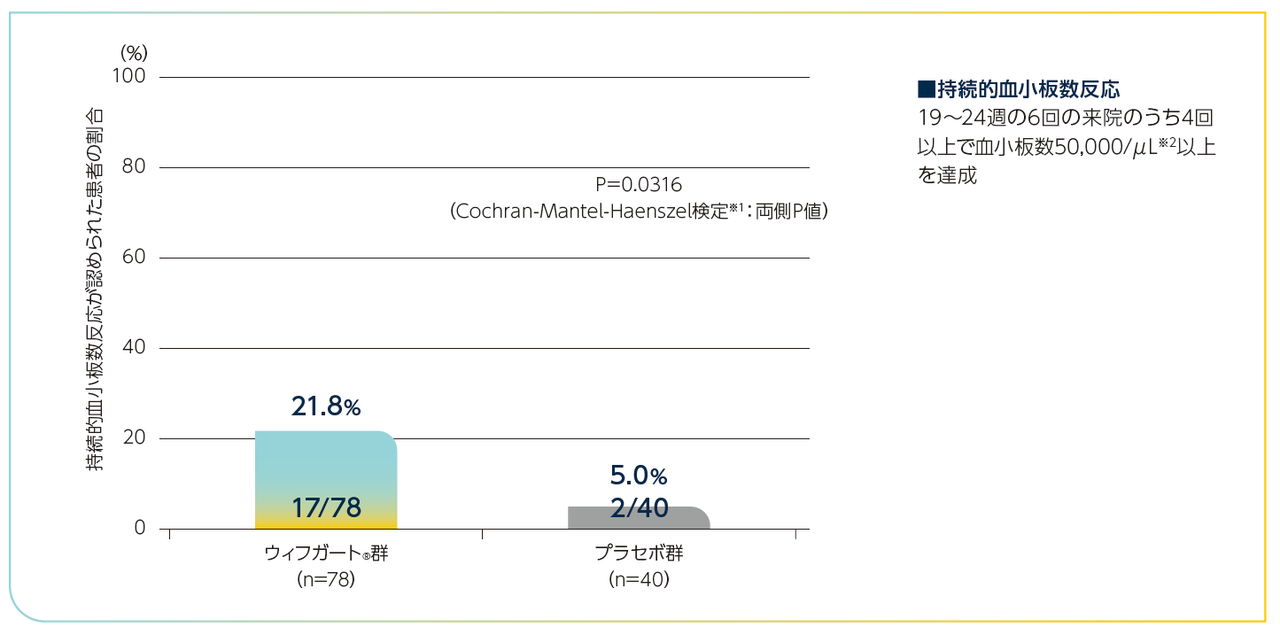
※1:中間事象(有効性の欠如又は有害事象による24週間の治療期間中の治験薬の投与中止、及び投与12週後以降のレスキュー治療の実施もしくは併用ITP治療薬の増強)が発生した患者については、持続的血小板数反応の未達成例と判定された。なお、上記中間事象以外の事由による血小板数の欠測については、欠測時点の前後の評価時点がいずれも治療期間内、かつ両時点で血小板数が50,000/μL以上の場合は50,000/μL以上として補完、それ以外の場合は50,000/μL未満として補完した。
※2:ランダム化の層別因子(脾臓摘出術の既往及びベースラインの併用ITP治療薬)並びにベースラインの血小板数カテゴリー(15,000/μL未満vs 15,000/μL以上)で層別解析を行った。
主要な副次評価項目
全体集団における持続的血小板数反応※1が認められた患者の割合
ウィフガート®群がプラセボ群と比較して統計学的に有意に高く、ウィフガート®群が86例中22例(25.6%)、プラセボ群が45例中3例(6.7%)でした(P=0.0108、Cochran-Mantel-Haenszel検定※2:両側P値)。
持続的血小板数反応が認められた患者の割合(全体集団)
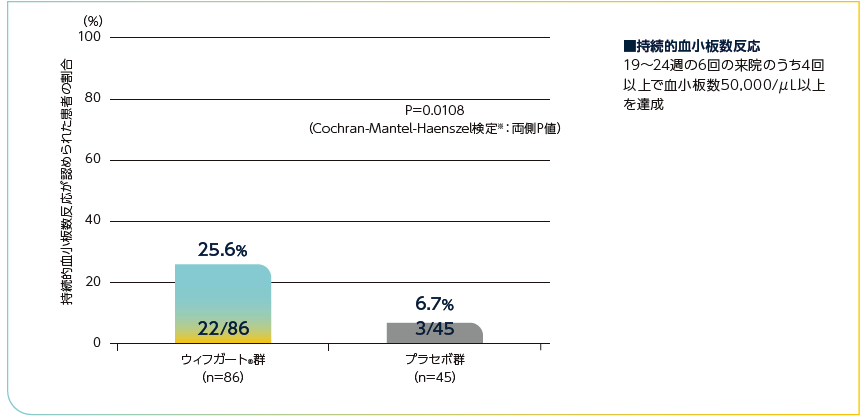
※1:中間事象(有効性の欠如又は有害事象による24週間の治療期間中の治験薬の投与中止、及び投与12週後以降のレスキュー治療の実施もしくは併用ITP治療薬の増強)が発生した患者については、持続的血小板数反応の未達成例と判定された。なお、上記中間事象以外の事由による血小板数の欠測については、欠測時点の前後の評価時点がいずれも治療期間内、かつ両時点で血小板数が50,000/μL以上の場合は50,000/μL以上として補完、それ以外の場合は50,000/μL未満として補完した。
※2:ランダム化の層別因子(脾臓摘出術の既往及びベースラインの併用ITP治療薬)並びにベースラインの血小板数カテゴリー(15,000/μL未満vs 15,000/μL以上)で層別解析を行った。
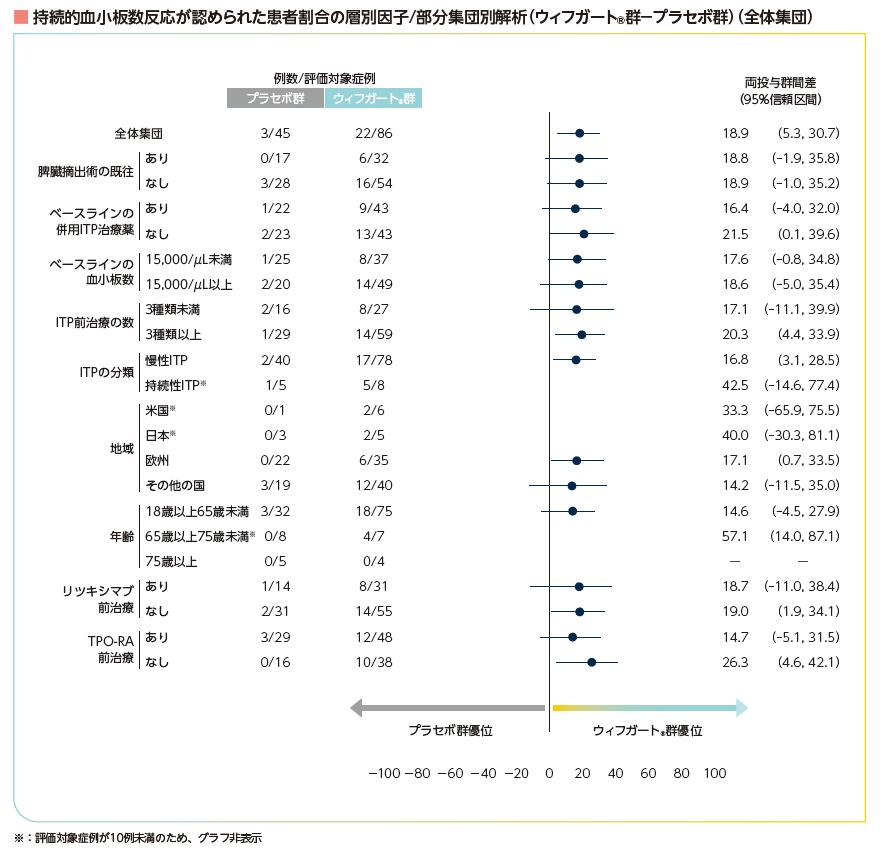
層別因子及び部分集団別の持続的血小板数反応(部分集団解析)4)
ランダム化の層別因子及び部分集団において、19~24週の6回の来院のうち4回以上で血小板数50,000/μL以上を達成した患者の割合の差及び95%信頼区間は下図の通りでした。
持続的血小板数反応が認められた患者割合の層別因子/部分集団別解析(ウィフガート®群-プラセボ群)(全体集団)
その他の副次評価項目
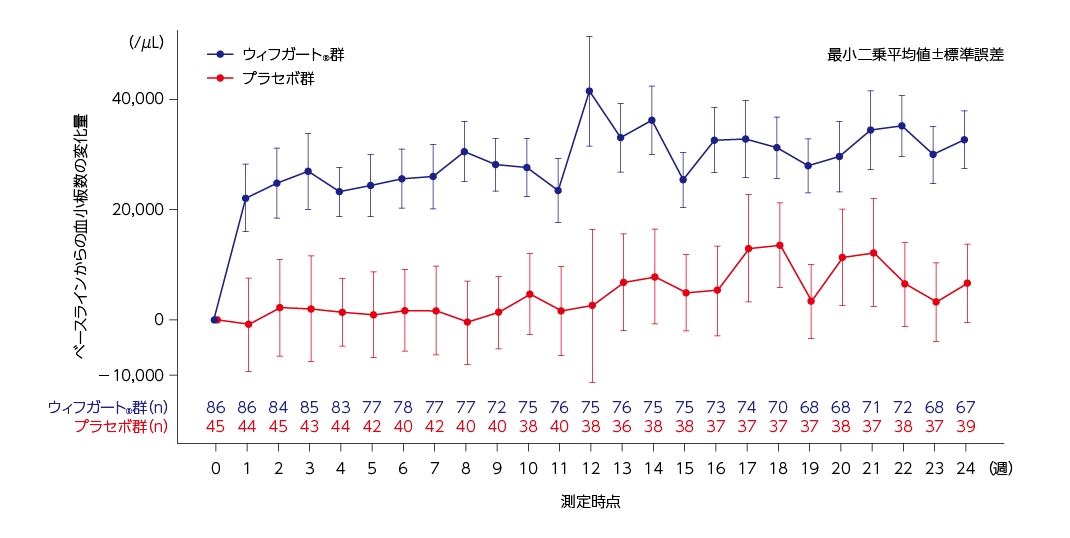
全体集団における各来院時の血小板数のベースラインからの平均変化量
混合モデルに基づく血小板数のベースラインからの平均変化量の推移は下図の通りでした。
MMRMに基づく血小板数の1週でのベースラインからの変化量の最小二乗平均値(標準誤差)は、ウィフガート®群が22,151(6,127)/μLに対して、プラセボ群は−894(8,494)/μLでした。
ベースラインからの血小板数の変化量(全体集団)2)
その他の副次評価項目
全体集団におけるレスキュー治療実施率
レスキュー治療を実施した患者の割合は、ウィフガート®群33.7%(29/86例)及びプラセボ群48.9%(22/45例)でした。その内訳は、副腎皮質ステロイド†が29.1%(25/86例)及び28.9%(13/45例)、免疫グロブリン大量療法が15.1%(13/86例)及び40.0%(18/45例)、血小板輸血が2.3%(2/86例)及び13.3%(6/45例)でした。
†:ITP治療に対し本邦未承認の薬剤を含む
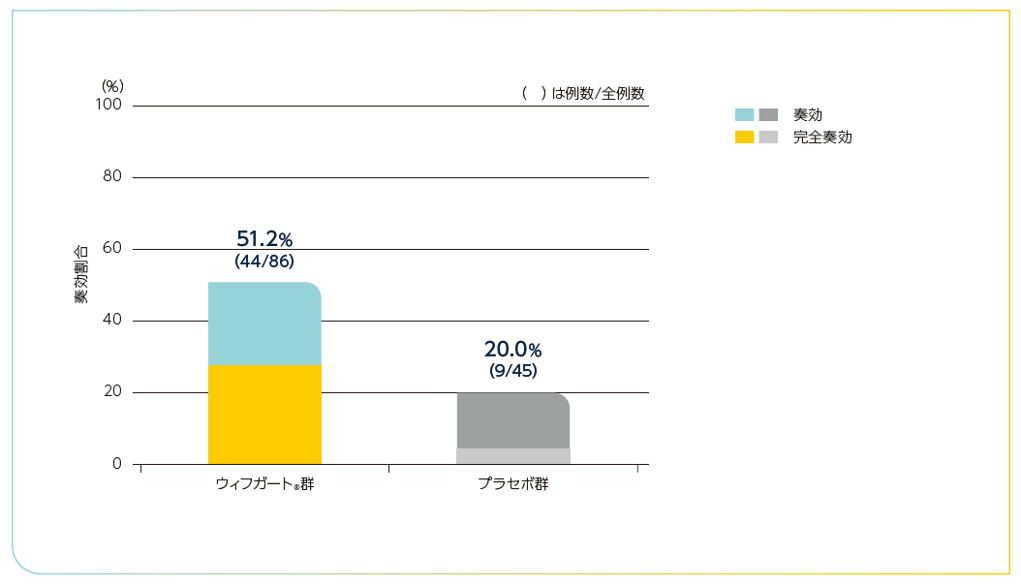
探索的評価項目
全体集団におけるIWGによる効果判定基準を満たした患者の割合2)
血小板数及び出血性イベントの両方を含むIWGによる効果判定基準を満たした患者の割合は以下の通りでした。
IWGによる効果判定基準を満たした患者の割合(全体集団)
| ウィフガート®群 (n=86) | プラセボ群 (n=45) | |
|---|---|---|
| IWG完全奏効 | 24(27.9%) | 2(4.4%) |
| IWG奏効 | 44(51.2%) | 9(20.0%) |
| IWG初回奏効 | 27(31.4%) | 3(6.7%) |
IWG効果判定基準による奏効割合
IWG基準
その他
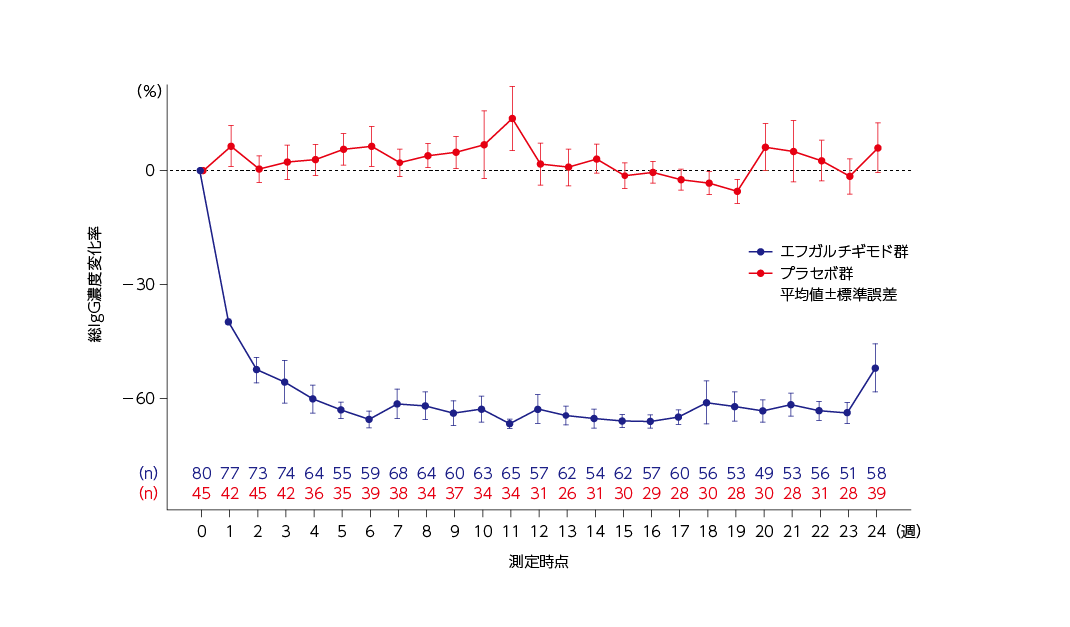
薬力学
総IgG濃度変化率の推移(全体集団)6)
ベースラインからの総IgG濃度変化率の推移は下図の通りでした。
総IgG濃度は、ウィフガート®の週1回投与開始後の最初の4週間を通して減少し、4週にはほぼ最大の減少に達しました。その後、総IgG濃度のベースラインからの平均減少率は、24週の治療薬投与期間を通して60%程度を維持しました。
投与頻度固定期間(16~23週)の総IgG濃度のベースラインからの平均減少率は、週1回投与及び2週に1回投与ともに60%程度を維持しました。
総IgG濃度のベースラインからの変化率推移(全体集団)
安全性評価
すべての有害事象(全体集団)
全体集団における有害事象はウィフガート®群で86例中80例(93.0%)、プラセボ群で45例中43例(95.6%)に認められました。
主な有害事象(各投与群の10%以上)
ウィフガート®群:尿中血陽性31例(36.0%)、挫傷17例(19.8%)、頭痛及び血尿各14例(16.3%)、点状出血13例(15.1%)
プラセボ群:尿中血陽性17例(37.8%)、点状出血12例(26.7%)、血腫11例(24.4%)、口腔内出血及び鼻出血各8例(17.8%)、血尿7例(15.6%)、歯肉出血、挫傷、頭痛及び斑状出血各6例(13.3%)、四肢痛5例(11.1%)
重篤な有害事象
ウィフガート®群で7例(8.1%)12件、プラセボ群で7例(15.6%)8件に認められました。
ウィフガート®群:血小板減少症2例3件、筋骨格系胸痛1例2件、鉄欠乏性貧血、腹痛、サイトメガロウイルス感染、丹毒、慢性骨髄単球性白血病、頭痛、膣出血各1例1件
プラセボ群:貧血、免疫性血小板減少症、口腔内出血、COVID-19、虫垂炎、交通事故、急性腎障害、血尿各1例1件
投与中止に至った有害事象
ウィフガート®群で4例(4.7%)4件、プラセボ群で1例(2.2%)1件に認められました。
ウィフガート®群では、血小板減少症、気管支炎、慢性骨髄単球性白血病、蕁麻疹でした。
プラセボ群では、COVID-19でした。
死亡に至った有害事象
本試験では死亡例は認められませんでした。
特に注目すべき有害事象(AESI)と定義した出血性事象
ウィフガート®群で61例(70.9%)277件、プラセボ群で39例(86.7%)237件に報告されました。
特に注目すべき有害事象(AESI)と定義した感染症
ウィフガート®群で25例(29.1%)37件、プラセボ群で10例(22.2%)12件に報告されました。
治験薬との因果関係が否定できないすべての有害事象(全体集団)
| 事象名 | ウィフガート®群 (n=86) | プラセボ群 (n=45) |
|---|---|---|
| 例数(%) | 例数(%) | |
| 発現例数(発現率) | 15(17.4) | 10(22.2) |
| 胃腸障害 | 1(1.2) | 2(4.4) |
| 下痢 | 1(1.2) | 0 |
| 悪心 | 0 | 1(2.2) |
| 嘔吐 | 0 | 1(2.2) |
| 一般・全身障害および投与部位の状態 | 5(5.8) | 0 |
| 無力症 | 1(1.2) | 0 |
| 悪寒 | 2(2.3) | 0 |
| 注射部位静脈炎 | 1(1.2) | 0 |
| 倦怠感 | 1(1.2) | 0 |
| 発熱 | 1(1.2) | 0 |
| 肝胆道系障害 | 1(1.2) | 0 |
| 肝毒性 | 1(1.2) | 0 |
| 免疫系障害 | 1(1.2) | 0 |
| 低γグロブリン血症 | 1(1.2) | 0 |
| 感染症および寄生虫症 | 2(2.3) | 1(2.2) |
| 口腔ヘルペス | 1(1.2) | 0 |
| 上気道感染 | 1(1.2) | 0 |
| 尿路感染 | 1(1.2) | 1(2.2) |
| 傷害、中毒および処置合併症 | 1(1.2) | 1(2.2) |
| 注入に伴う反応 | 1(1.2) | 0 |
| 処置によるめまい | 0 | 1(2.2) |
| 臨床検査 | 0 | 1(2.2) |
| 血中尿酸増加 | 0 | 1(2.2) |
| 代謝および栄養障害 | 1(1.2) | 0 |
| 高尿酸血症 | 1(1.2) | 0 |
| 低カリウム血症 | 1(1.2) | 0 |
| 筋骨格系および結合組織障害 | 2(2.3) | 1(2.2) |
| 関節痛 | 1(1.2) | 0 |
| 側腹部痛 | 1(1.2) | 0 |
| 四肢痛 | 0 | 1(2.2) |
| 神経系障害 | 3(3.5) | 4(8.9) |
| 頭痛 | 3(3.5) | 3(6.7) |
| 錯感覚 | 0 | 1(2.2) |
| 呼吸器、胸郭および縦隔障害 | 1(1.2) | 0 |
| 咳嗽 | 1(1.2) | 0 |
| 皮膚および皮下組織障害 | 3(3.5) | 2(4.4) |
| そう痒症 | 2(2.3) | 1(2.2) |
| 斑状丘疹状皮疹 | 0 | 1(2.2) |
| 蕁麻疹 | 1(1.2) | 0 |
| 血管障害 | 0 | 1(2.2) |
| ほてり | 0 | 1(2.2) |
MedDRA version 24.1
1)社内資料:第3相試験ARGX-113-1801試験(承認時評価資料)(CTD2.7.6.3)(EFG90103)
2)社内資料:1801試験(第3相)(承認時評価資料)(CTD2.7.3.2.1)(EFG90095)
3)社内資料:試験対象集団(承認時評価資料)(CTD2.7.3.3.1)(EFG90098)
4)社内資料:部分集団における結果の比較(承認時評価資料)(CTD2.7.3.3.3)(EFG90098)
5)Broome CM, et al.: Lancet. 2023; 402(10413); 1648-1659. (EFG00139)
[COI] 本試験はargenx社の支援のもと行われた。著者にargenx社より講演料、コンサルタント料等を受領している者が含まれる。
また、著者にargenx社の社員が含まれる。
6)社内資料:薬力学的作用(承認時評価資料)(CTD2.7.2.2.2)(EFG90089)
おすすめコンテンツ
JP-VJITP-25-00537(2025年11月作成)
